 Gymnasium Ulricianum Aurich Europaschule
Gymnasium Ulricianum Aurich Europaschule
Vom 08. bis zum 19. Oktober wurde uns, Carina Geyken und Marlene Dirks, durch die Auricher Wissenschaftstage ein Praktikum am Max-Planck-Institut für Immunbiologie und Epigenetik in Freiburg, genauer gesagt im Labor von Prof. Dr. Thomas Jenuwein, ermöglicht.
Das Labor, in dem wir zwei Wochen lang mitgearbeitet haben, forscht im Bereich der Epigenetik. Epigenetik bezeichnet die Wissenschaft, die sich mit Veränderungen des Chromatins beschäftigt, einer Funktionseinheit, die aus Proteinen und der sich darum wickelnden DNA besteht. Das Besondere daran ist, dass die DNA, die als Träger sämtlicher genetischer Information bekannt ist, dadurch nicht verändert wird und jene Veränderungen am Chromatin dennoch vererbbar sein können. Außerdem spielen Umwelteinflüsse eine Rolle in der Epigenetik. Sie waren jedoch kein Thema während unseres Praktikums.
In der ersten Woche wurden wir von Dr. Nicholas Shukeir betreut und beschäftigten uns mit zwei essentiellen Methoden des Laboralltags: Immunfluoreszenz und Western Blot.
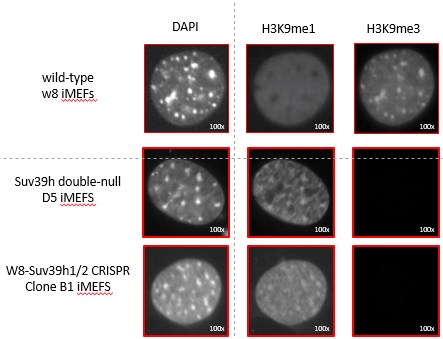
Bei einer Immunfluoreszenz werden
mithilfe von fluoreszierenden Antikörpern Gene sichtbar gemacht, die dann unter
einem Mikroskop untersucht werden können. Außerdem gibt die Immunfluoreszenz
Auskunft über den Ort der Gene. Dieses Prinzip haben wir am Beispiel von
Methyl-Markierungen an Histonen (den Proteinen, die von der DNA umwickelt
werden) von verschiedenen Mauszelltypen ausprobiert.
Dazu müssen die Zellen zunächst auf sogenannte „slides“ transferiert werden.
Bei den „slides“ handelt es sich um Glasplatten mit acht Unterteilungen, die jeweils
ein Volumen von etwa zwei Milliliter enthalten können.
Um später ein deutliches Bild unter dem Mikroskop sehen zu können, müssen die
Zellen zuerst fixiert werden. Dies geschieht durch die Chemikalie PFA, welche
im Puffer PBS gelöst ist. Danach haben wir mithilfe eines Toxins die
Zellmembranen der Mauszellen permeabilisiert, also im Prinzip winzige Löcher in
der Membran induziert. Diese sind im nächsten Schritt von großer Bedeutung, wenn
zwei verschiedene primäre Antikörper inkubiert werden, die entweder an die
Methyl-Markierung H3K9me1 oder H3K9me3 andocken sollen. Die Antikörper würden
ohne die Permeabilisierung der Zellmembran nur schlecht in die Zellen gelangen
und so das Mikroskopiebild negativ beeinflussen. Nach dem Inkubieren der
primären Antikörper werden sekundäre Antikörper inkubiert, die an die primären
Antikörper andocken sollen. Das hat zur Folge, dass die Antikörper insgesamt
eine größere Oberfläche haben und somit besser unter dem Mikroskop zu sehen
sind. Anschließend werden die Zellen und die darin enthaltenden Antikörper
erneut fixiert und dann mit DAPI, einem DNA-Farbstoff, eingefärbt. Im Anschluss
daran werden die „slides“ so bearbeitet, dass sie unter ein Mikroskop passen. Im
Folgenden kann unter dem Mikroskop untersucht werden, wo sich die DNA und die
H3K9me1- beziehungsweise H3K9me3-Markierungen befinden. Unter dem Mikroskop
lässt sich als Ergebnis feststellen, dass alle Markierungen im Zellkern
auftreten.
In den letzten Tagen der ersten Woche haben wir uns dann mit der Methode des Western Blots vertraut gemacht. Western Blot bedeutet, dass verschiedene Substrate über ein Gel laufen und dann deren Proteine sichtbar und hinsichtlich ihrer Masse und Menge vergleichbar gemacht werden. Als Ausgangspunkt dienten uns die gleichen Zellkulturen wie bei der Immunfluoreszenz.
Die Zellen werden bei der Immunfluoreszenz im ersten Schritt lysiert, was bedeutet, dass die Membran der Zellen aufgelöst wird. Dies geschieht durch einen RIPA-Puffer, dessen Wirkung durch Schallwellen verstärkt wird. Um ausschließlich die Proteine weiterverwenden zu können, wird das Produkt zentrifugiert. Dabei entsteht ein festes „pellet“ und eine Flüssigkeit. Für das Western Blot wird nur die proteinhaltige Flüssigkeit genutzt. Die Flüssigkeiten der verschiedenen Zellen werden dann in eine Gelkammer gefüllt, welche daraufhin an einen Stromkreis angeschlossen werden. Die Flüssigkeiten laufen ihrem Gewicht entsprechend das Gel hinunter. Dadurch entstehen Markierungen, die im Anschluss auf eine papierähnliche Membran übertragen werden, welche danach, vergleichbar mit der Immunfluoreszenz, mit Antikörpern behandelt wird. Diese Membran kann anschließend auf eine Folie kopiert werden, um die Ergebnisse festzuhalten. Dabei gilt, je dicker die Bande ist, desto mehr Protein ist in ihr enthalten. Außerdem bedeutet die Beobachtung, Banden auf der gleichen Höhe, dass sich das gleiche Protein im Ausgangstoff befindet.

Während der zweiten Woche wurden wir von Dr. Lisa Jerabek begleitet, die mit uns Proteine aufgereinigt und untersucht hat und uns außerdem ihre aktuellen Projekte nähergebracht hat.
Um Proteine zu reinigen, müssen natürlich erst Proteine entstehen. Dazu haben wir E. coli Bakterien genutzt, in die wir IPTG hinzugefügt haben, welches dafür sorgt, dass unser „Zielprotein“ von den Bakterien synthetisiert wird. Dann werden die gelösten Bakterien zentrifugiert, damit sie sich unten im Gefäß in Form eines „pellets“ absetzen. Dieses „pellet“ wird danach resuspendiert, also wieder aufgelöst. Durch Schallwellen wird die Zellmembran zerstört und nach erneuter Zentrifugation lassen sich die Proteine extrahieren. Im folgenden Schritt werden die Proteine gereinigt, indem sie durch eine Maschine mit einer sogenannten MBP-Säule laufen, die dafür sorgt, dass sich die Proteine daran binden. Die Säule wird danach mehrmals mit Maltose ausgewaschen, wobei sich die Proteine darin lösen. Auf diese Weise entstehen mehrere Lösungen mit unterschiedlichen Proteinkonzentrationen. Diese Lösungen werden dann auf ein Gel geladen, wodurch die Proteinkonzentration geschätzt werden kann. Die Lösung mit der höchsten Konzentration wird dann weiterverwendet. An den Proteinen hängt noch ein „tag“, der nun von dem Protein getrennt wird. Die übrigen Proteine werden erneut über eine Säule, diesmal eine His-Säule, gewaschen. Anschließend werden die Proteine erneut auf ein Gel geladen. Außerdem wird ein „Vergleichsprotein“ dazu geladen, mit dessen Hilfe sich dann einschätzen lässt, wie groß die Konzentrationen sind. Als letztes wird die sogenannte EMSA durchgeführt, die die mögliche Bindung von unseren Proteinen an doppelsträngige DNA, doppelsträngige RNA und/oder DNA:RNA-Hybride nachweisen kann. Für die EMSA sind besondere Gele notwendig, die wir am Vortag selbst hergestellt haben.

Carina Geyken beim Gießen der EMSA-Gele.

Marlene Dirks beim Laden der EMSA-Gele.

Am Wochenende zwischen den beiden Praktikumswochen haben wir unsere freie Zeit genutzt, indem wir uns Fahrräder ausgeliehen haben und damit nach Frankreich gefahren sind. Dort haben wir die Stadt Neuf-Brisach besichtigt, die sich durch ihre beeindruckende Festungsanlage auszeichnet. Am Sonntag machten wir uns dann auf den Weg zu einer weiteren, eher lokalen Sehenswürdigkeit, der Zähringer Burgruine, deren Turm eine grandiose Aussicht über Freiburg bietet. Unter der Woche haben wir gelegentlich die Altstadt Freiburgs besucht und Bereiche des nahegelegenen Waldes erkundet.

Die grandiose Aussicht vom Turm der Zähringer Burgruine.
Carina Geyken und Marlene Dirks

Marlene Dirks und Carina Geyken vor dem Haupteingang des Instituts.

Unser Arbeitsplatz, auch Bay genannt.
